
AiPE
[기타] 컴퓨터는 분자구조를 어떻게 계산할까 본문
고등학교 2학년 화학Ⅰ과목 발표 시간에 사용했던 발표자료이다.
마찬가지로 고치기 귀찮아서 발표대본을 그냥 올린다.
Ⅰ. 서론
현재 대학 과정의 화학은 실험화학과 이론화학의 두 가지 분야로 나뉜다. 보통 실험실에서 다양한 약품을 섞고 가공하는 모습은 실험화학 과정의 모습인데, 이론화학의 한 분야인 계산화학은 물질을 직접 만들고, 다루고, 관찰하는 실험화학과는 달리 종이와 펜, 컴퓨터만으로 실험이 진행된다. 실험화학은 다수의 실험 사실로부터 그 뒤에 있는 보편적인 이로을 끌어냄으로서 귀납적이지만, 계산화학을 포함한 이론화학은 이론적인 모델과 수식을 바탕으로 기존의 현상을 증명하거나 미지의 물질의 성질을 예측하는 연역적인 분야라고 볼 수 있다.
Ⅱ. 컴퓨터 소프트웨어상 분자 구조 결정의 방법
이러한 계산화학에서 가장 사랑받는 도구는 단연 컴퓨터인데, 컴퓨터는 다음과 같은 방법을 통해 분자구조를 결정한다.

글 내의 모든 사진 출처 : (윤정혁, 노경태, 1998) 컴퓨터 분자설계방법을 이용한 고분자성 물질의 예측
컴퓨터가 분자구조를 결정하는 방식은 크게 [이론적 방법]과 [Hybrid 방법]으로 나뉜다. [이론적 방법]은 분자를 이루는 원자와 그 전자들의 반발 및 이동을 직접 계산하여 분자 구조를 결정하는 방식이고, [Hybrid 방법]은 기하학적인 원리 등을 이용해 분자 구조를 결정하는 방식이다.
이론적 방법은 [양자역학적 방법]과 [분자역학적 방법]으로 나눌 수 있다. 용어 설정의 오류로 인해 [이론적 방법]은 넓은 범위에서는 [양자역학적 방법]과 [분자역학적 방법] 모두를 포함하기도, [양자역학적 방법]만을 포함하는 의미로 쓰이기도 한다.
[이론적 방법]이 좁은 의미로 쓰일 경우 [분자역학적 방법]은 [경험적 방법]이라는 명칭을 부여받는데, 본 소논문에서는 용어의 혼선을 방지하기 위해 [이론적 방법]을 큰 의미로 보고 [양자역학적 방법]과 [분자역학적 방법]이라는 용어를 사용하겠다. 또한 본 소논문에서는 [Hybrid 방법]에 대한 이해가 부족한 관계로 [이론적 방법]에 대해서만 다루겠다.
*이 포스팅의 목차와 글 구조도 위 트리 구조를 그대로 따라갑니다. 분류가 복잡하고 많아서 읽다 보면 헷갈릴텐데 위 사진의 트리 구조를 보고 현재의 방법이 어디에 속해있는 무슨 방법인지 이해해주세요.
Ⅲ. 양자역학적 방법
[양자역학적 방법]은 슈뢰딩거 방정식을 푸는 것으로부터 시작한다. 시간 비의존 슈뢰딩거 방정식을 정리한 식인 \(H psi (r)=E psi (r)\)에서 \(H\)는 \(Hamiltonian\), \(E\)는 에너지, \(psi\)(psi, 사이)는 파동함수를 나타낸다.
위 개념들에 대한 정의를 알고싶다면 아래 문서를 참고하자. 나도 발표 준비를 하면서 아래 문서를 참고했다.
https://namu.wiki/w/%EC%8A%88%EB%A2%B0%EB%94%A9%EA%B1%B0%20%EB%B0%A9%EC%A0%95%EC%8B%9D
슈뢰딩거 방정식 - 나무위키
해밀토니언(H\mathcal{H}H)이 시간 ttt에 의존하지 않는 경우[6](ttt에 대한 함수가 아닌 경우) 슈뢰딩거 방정식은 다음과 같이 간단한 꼴로 쓸 수 있다. E∣ψ>=H^∣ψ> E \left|\psi\right>= \hat{\mathcal{H}} \left|\
namu.wiki
슈뢰딩거 방정식은 양자역학적 관점에서 물질의 상태를 기술하는 방정식으로, 시간 의존 슈뢰딩거 방정식과 시간 비의존 슈뢰딩거 방정식으로 나눌 수 있다. 이 둘은 슈뢰딩거 방정식의 해밀토니언\((H)\)가 시간변수\((t)\)에 대한 의존 여부가 다르다.
여기서 해밀토니언\((H)\)에 대한 간단한 이해가 필요하다. 해밀토니언은 일반 좌표계에서의 일반화 운동량이다. 즉, 어떤 시스템의 \(E_k\)와 \(E_p\)의 총합이 곧 해밀토니언이 된다고 이해하면 편리하다. (자세한 정의는 위 나무위키 문서를 참고하자.) 단, 이 정의가 만족하기 위해서는 다음 두 조건을 만족시켜야 한다.
| 1. \(E_p\)는 속도 \(x_a,i\)와 의존하지 않아야 한다. |
| 2. 입자의 좌표 \(x_a.i\)와 일반화 좌표 \(q_i\)간의 변환식이 시간에 의존하지 않아야 한다. |
위의 방정식을 푸는 계산을 통해 \(psi(r)\)을 구하고 이를 이용하여 각종 분자의 성질을 얻는다는 컨셉이 [양자역학적 방법]의 기본이다.
[양자역학적 방법]은 이론적인 접근방법 중 정확성은 가장 높지만 정확하게 계산할 수 있는 분자의 크기가 제한되어 있다는 한계가 있다.
Ⅲ-1. Ab initio 방법 (Ab initio quantum chemistry methods)
순수 분석적 함수를 이용하여 계산하는 방법을 Ab initio 방법이라고 한다. 다른 원칭은 [제 1 원리 계산]이며, Ab initio라는 말은 라틴어로 “from the beginnng”이라는 뜻을 가진다. 계산이 다른 경험적 수량을 전혀 사용하지 않았다는 의미이다. 즉, 아무런 데이터가 없어도 계산만으로 분자의 구조를 비교적 정확히 결정할 수 있다.
이론상으로, Ab initio방법에서는 슈뢰딩거 방정식을 풀 수 있다는 전제 하에, 양성자, 중성자 및 전자의 수와 확률적 위치만 알면 우리는 그 계의 모든 정보를 알 수 있다.
그러나 원자와 전자의 수가 많은 계에서는 슈뢰딩거 방정식을 완벽하게 풀기란 거의 불가능에 가깝다. 따라서 원자와 전자의 수많은 상호작용과 수많은 포텐셜 변화 등으로 인해서 적당한 가정이 필요하게 된다. 거의 안 움직이는 입자는 안 움직인다고 가정하고나 상호작용이 미미한 경우 그냥 없다고 가정하는 방식이다. 이를 근사(Approximation)이라고 한다.
#1. Hartree-Fock 근사 (HF)
전자에 비해 매우 무거운 원자를 움직이지 않는다고 가정 (Born-Oppenheimer 근사) 하고, 전자와 전자의 상호작용이 고려되지 않으며 각각의 전자를 평균 내어 동일하게 취급 (Mean-field 근사) 하여 변분법을 통해 수렴값을 찾는 방식이다. 파동 함수에서 얻어진 계의 에너지가 실제와 같거나 다소 크게 계산되는 필연적인 오차(HF 한계)를 가진다.
#2. Hartree-Fock 이후의 방법 (Post HF)
대부분 HF 근사법에서는 다루지 않고 있는 전자와 전자 사이의 상호작용을 보정하기 위해 추가적인 항을 도입한 방법들입니다. 보통 HF 근사법보다 정확하지만, 계에 따라서는 그렇지 않은 경우도 존재한다.
| 1) 묄러-플레셋 섭동이론, Møller–Plesset perturbation theory, MP 전자 사이의 상호작용은 레일리-슈뢰딩거 섭동이론 (Rayleigh–Schrödinger perturbation theory)을 통해 기술하며, 섭동 함수의 차수에 따라 2차(MP2), 3차(MP3), 4차(MP4)로 나누어 사용한다. 2) 배열 상호작용, Configuration interaction, CI 전자 사이의 상호작용과 들뜸을 배열 상태함수 (Configuration state functions, CSFs) 라는 변분법적 파동함수를 도입하여 계산한 방식이다. 얼마나 많은 숫자의 스핀 궤도함수 (spin orbitals, SO) 들을 고려하였는가에 따라 행렬식의 수가 달라지며 한 개만 고려한 CIS, 두 개만 고려한 CID, 하나의 두 개의 들뜸을 모두 고려한 CISD 등이 있다. 3) 짝지어진 클러스터, Coupled cluster, CC exponential cluster operator 를 사용한 다전자 wavefunction 을 도입하여 계산한 방식이다. 슬레이터 행렬식의 linear combination 을 사용한 CI 에 비해 exponential term 이 추가되어 더 정확한 결과를 얻을 수 있으며, 이 또한 몇 개의 들뜸을 고려하였는가에 따라 CCS, CCSD, CCSDT 와 같이 분류할 수 있다. CCSDT의 경우 들뜬 상태를 계산하는 가장 정확한 방법 중 하나로 사용하기도 한다. 4) 그 외 Quadratic configuration interaction (QCI), Quantum chemistry composite methods 및 들뜬 상태와 바닥 상태의 상호작용까지 고려한 다양한 다중참조 방법 (MCSCF, MRCI, NEVPT, CASPTn) 들이 존재한다. |
Ⅲ-2. 반경험적 방법 (Semi-emprical Method)
Ab initio의 계산을 위해 시간 비의존 슈뢰딩거 방정식 \(H psi (r)=E psi (r)\)을 풀 때, 일부의 계산은 계산시간 단축을 위해 실험에서 얻어지는 매개변수로 대치하여 계산하는 방법을 반경험적 방법이라고 한다.
즉, 기본적인 원리는 양자의 직접적인 “수치 계산”을 통한 분자구조 결정이지만 이 수치를 구하는 방법에 있어 기존의 구해놓은 값들을 응용한다는 점에서 차이가 있다.
Ⅲ-3. 범밀도함수법 (Density function Method)
가장 널리 쓰이는 양자계산 분야 중 하나이다. 전공생들이 가장 처음으로 접하는 이론이라고 하는데, 파동함수를 도출하는 HF 및 post-HF 계산법과는 달리, 전자들의 파동함수 대신 파동함수를 제곱한 밀도함수로 대체해 계산함으로서 복잡도를 줄이는 방식이다. 표준편차를 구하기 위해 복잡한 루트 계산을 수행하는 대신 분산을 구해 루트를 씌우는 것과 같은 맥락으로 이해하면 편할 듯 하다.
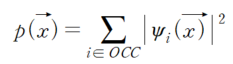
공간상의 어떤 점에 전자가 존재할 때, 이 전자가 느끼는 \(E_p)\)는 다른 전자들의 밀도함수를 통해 평균장(mean field) 을 계산함으로써 정의할 수 있으며 HF 에 비해 매우 빠르고 효율적인 계산을 가능하게 한다. 속도와 효율성 덕분에 실용적인 분야에서 가장 널리 쓰이는 계산법이지만 전하 이동이 큰 계에서는 정확도가 떨어진다는 단점이 존재한다.
따라서 HF 계산과 혼합하여 사용하는 hybrid DFT, 들뜬 상태를 계산하기 위한 Time dependent DFT (TD-DFT) 등 여러 방법이 개발되어 있다. 이들이 맨 처음 소개한 [hybrid 방법]의 일종이다.
Ⅳ. 분자역학적 방법
큰 분자의 구조 계산이 가능하도록 원자들 사이의 상호작용을 묘사할 수 있는 (계산보다는 묘사라는 표현이 적절하다) 간단한 함수를 실험 데이터나 양자계산 결과를 이용하여 ‘결정’한다. 이 과정에서 각 원자 또는 원자 쌍에 적합한 매개변수가 할당되며 이 함수들을 통해 분자간의 상호작용을 설명하게 된다.
보통 이 때 얻어지는 함수를 힘장(force field)이라고 한다. 각 원자에 할당되는 매개변수는 전하, \(vander Waals\) 반지름 등 여러 종류가 있다.
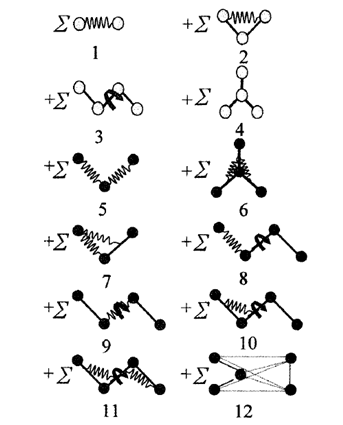
분자 내 분자 간 상호작용 에너지(\(E\))는 힘장을 통해 계산되며 보통 힘장은 다음 그림과 같은 여러 가지 분자운동에 해당되는 \(E_p\) 항들로 구성된다.
따라서 전체 힘장의 구성 성분은 아래와 같다.

Ⅳ-1. 에너지 최소화법 (Energy Minimization)
분자의 에너지는 분자를 이루는 원자의 위치에 대한 함수로 계산할 수 있다. 단분자 또는 분자계는 열역학 법칙에 따라 에너지가 가장 낮은 상태를 유지하려고 하는데, 이때 안정한 구조를 가지기 위한 각 분자의 에너지를 계산하는 방법이다.
 |
분자의 전체 에너지가 가장 낮은 상태에서는 원자의 위치(\(Δr\)) 변화에 따른 분자의 에너지 변화가 0이다. 따라서 \( \frac{dE}{dr}=0 \)을 만족하는 분자구조를 찾아 가장 안정한 구조를 결정할 수 있다. 구성원자의 위치변화에 따른 에너지는 아래와 같이 표현된다.
$$E(r+\Delta r)=E(r)+\frac{dE}{dr}\bullet \Delta r$$
컴퓨터는 이 수식에서 \( \frac{dE}{dr}=0 \)인 \(r\)값을 결정하기 위한 알고리즘을 이용하는데, 이 알고리즘은 Steepest descent, Conjugate gradient, Newton-Raphson등이 있다. 다른 알고리즘들은 이 세 알고리즘에서 변형된 것들이 많다.
이때, 두 번 이상 미분한 식은 에너지가 너무 작아 실질적인 계산에 방해만 될 뿐이므로 0으로 근사하여 생략하겠다. 이를 정리하면 다음과 같은 일차 근사식이 나온다.

Ⅳ-2. 몬테카를로 방법 (Monte Carlo Method)
몬테칼로 방법은 통계적인 방법으로써 가중치 평균을 이용해 분자구조\((r)\)에 대한 정보나 열역학적인 성질을 얻는 방법인다. 이를 수식으로 표현하면 다음과 같다.

위의 계산은 주로 Metropolis 알고리즘을 사용한다. 알고리즘의 큰 틀은 아래와 같다.

Ⅳ-3. 분자동력학 (Molecular Dynamics)
분자동력학 방법은 결정론적인 과정(causility process)에 의해 그 분자가 존재 가능한 구조들을 시간과 위치에 대한 함수로 표현한다. 식 자체는 에너지최소화법에서 이용했던 식과 동일하다.
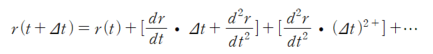
이 또한 에너지 최소화법에서와 같은 이유로 근사하여 이차항까지만 나타내겠다.

이 식에서 \(\frac{dr}{dt}\)는 입자의 속도에 해당하며, \(\frac{d^2r}{dt^2}\)은 가속도를 나타낸다. 여기서 고전역학의 컨셉이 필요한데, 고전역학에서 포텐셜에너지함수\((E)\)를 1차미분하여 힘\(F)\)을 얻을 수 있다는 점을 이용하겠다.
뉴턴 운동 방정식 \(F = ma\)에서 \(a = \frac{dE(r)}{dr}\)을 대입하면 \(a = \frac{d^2r}{dt^2}\)이므로 \(F = m \bullet \frac{d^2r}{dt^2}\)이다. 이 식으로부터 원자가 받는 \(F\)를 계산할 수 있다. 앞서 근사한 식을 다시 표현하면 다음과 같다.

위 식에 따라 분자는 실제 존재 가능한 구조들을 가지게 되고, 이 과정을 통해 시간에 따른 분자의 궤적을 분석하면 분자구조에 대한 정보와 분자의 여러 가지 열역학적인 성질을 이해할 수 있다.
Ⅴ. 결론
컴퓨터가 분자 구조를 계산하며 결정하는 데에는 위와 같은 방법들이 범용된다. 또한 컴퓨터의 연산 능력은 우리가 생각하는 것 이상으로 빠르다. 따라서 복잡한 알고리즘을 사용하지 않아도 계산 능력을 통해 분자 구조를 결정할수 있다.
현재 컴퓨터에 사용되는 알고리즘은 Ab initio를 기반으로 하여 계산된 다양한 결과물들을 미리 저장해놓았다가 연산이 필요할 때 그 소스로서 사용하는 방식이 대다수이다. Ab initio의 특성상 계산할 수 있는 한 분자를 이루는 원자의 개수가 제한되어있기 때문에 큰 분자의 경우 분자를 여러 부분으로 잘라 Ab initio를 통해 계산한 뒤 다시 그들을 [분자역학적 방법]등으로 이어 붙이는 방식을 채택한다.
앞으로 양자 컴퓨터등의 발달로 인해 컴퓨터의 연산 능력이 비약적으로 증가하게 된다면 현재의 알고리즘만으로도 분자구조의 결정 또한 더 빠르고 정확해질 수 있을 것이다.
| 느낀 점 및 소감 |
| 원래는 계산화학에 대해 살짝만 발표할 생각이었는데, 하다보니까 재미있어서 너무 깊이 와버린 것 같다. 미적분을 배우고 나니 화학 수업 시간에 배웠던 것들을 색다른 방법으로 해석할 수 있게 되니 화학에 대한 흥미가 더 생긴 것 같다. 이번 발표에서는 계산화학의 큰 틀과 기본적인 알고리즘을 설명하는데 시간을 다 쓴 것 같은데, 눈에 보여지는 것들이 적어 아쉬웠다. 다음에 비슷한 주제로 또 발표를 진행하게 된다면 파이썬을 통해 직접 코딩을 한 뒤 비교적 값이 정확하게 나오는 플루오린분자(74nm)의 결합거리를 Metropolis 알고리즘을 통해 계산해보는 활동을 해 봐도 재미있고 유익할 듯 하다. 또한 계산화학의 양자역학적 방법을 공부하다보니 Ab initio또한 시간 비의존 슈뢰딩거 방정식을 근간으로 한다는 것을 깨닫게 되어 화학과 물리는 결국 뗄 수 없다는 것을 느끼게 되었다. 이번 발표를 통해 계산화학이라는 새로운 재미있는 분야를 알게 된 것 같아 보람차다. |
아래는 발표대본의 .hwpx파일이다.
내용은 위 글의 내용과 완전히 일치한다.
그리고 블로그 스킨에 다크모드를 키면 Ⅲ-1. Ab initio 부터 제목 색깔이 어둡게 보이는 현상이 있던데 아직까지 왜 그런지 모르겠다. 조만간 고쳐보겠다.
'Development Project > Etc.' 카테고리의 다른 글
| Python Pyinstaller enum34 오류 해결 (0) | 2023.02.07 |
|---|---|
| [학교 과제] 파이썬 Matplotlib으로 정규분포 곡선 (PDF, CDF) 그리기 (0) | 2023.02.06 |
| [Python] 특정 문자 2개 사이의 문자를 추출하는 법 (0) | 2023.01.29 |
| Python 패키지 Pyinstaller로 실행파일(.exe) 만드는 법 (2) | 2023.01.29 |
| [Python] "pyinstaller은(는) 내부 또는 외부 명령 실행할 수 있는 프로그램 또는 배치 파일이 아닙니다" 해결 (0) | 2022.12.10 |
